- 1
- 2
- 3
一、概述
ISO13485是用适用于医疗器械法规环境下的质量管理体系标准,其全称是《医疗器械 质量管理体系 用于法规的要求》。虽然ISO 13485是基于ISO 9001的计划、执行、检查、行动(PDCA)的流程模型理念,但其目的是确保医疗器械的合规。
因此,ISO13485更有针对性,并且对质量管理体系提出了更高的文件化的要求。ISO 13485旨在帮助医疗器械制造商建立质量管理体系,并维持体系的有效性。它能确保医疗器械在设计、开发、生产、组装、运输、处置等所有阶段的安全性并符合预期用途。
目前组织可以依据ISO13485:2016版标准建立体系或者寻求认证。
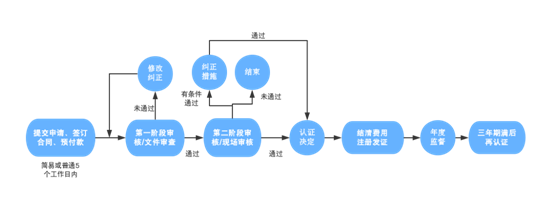
二、认证流程
管理体系相关认证的流程基本一致,企业申请时不需要特别记录。流程一般包括:提交申请、签订合同和交预付款;初审(第一阶段审核/文件审查,第二阶段审核/现场审核);认证决定;结算费用,注册发证;每年的监督审核(次数略有不同);证书期满后的再认证等环节。
如图所示:
三、申请条件
申请人应具有明确的法律地位;
申请人应具备相应的许可资质:
1.对于生产型企业,I类产品需提供医疗器械产品备案凭证以及生产备案凭证;II类及III类产品需提供医疗器械产品注册证和医疗器械生产企业许可证;
2.对于经营企业,经营II类产品的需要提供医疗器械经营企业备案凭证;经营III类产品的需要提供医疗器械经营企业许可证;
3.对于仅出口的企业,根据3月31日商务部、海关及药监局三部委的文件,出口医疗防疫物品在满足进口国要求的前提下还需要取得国内医疗器械产品注册证/备案凭证以及医疗器械生产企业许可证/备案凭证;
4.申请人已经按照标准建立文件化的管理体系(包括质量手册、程序文件、内审资料、管理评审资料以及程序文件要求的其它相关表单);
认证申请前,管理体系至少有效运行三个月并进行了一次完整的内部审核和管理评审(对于生产植入性医疗器械产品,体系运行时间至少6个月,其他产品的管理体系至少运行3个月)。
四、认证范围
根据IAF MD 9:2017《强制性文件ISO/IEC 17021 应用于ISO13485 医疗器械质理管理体系》要求,并参考中国大陆地区《医疗器械分类目录》编制。
ISO13485认证范围分类
非有源医疗设备;
有源(非植入)医疗器械;
有源(植入)医疗器械;
对医疗器械的灭菌方法;
体外诊断医疗器械;
医疗器械有关服务;
1.受审核组织认证范围必须是在UKAS认可的范围内;
2.归属于国内食药局发布的《医疗器械分类目录》内的认证产品,需提供国内医疗器械的产品/生产/经营备案凭证或注册证,方可受理;
3.不属于国家药监局发布的《医疗器械分类目录》内,且产品仅供外销的,能提供在相应的出口国按照医疗器械管理的证明(例如出口欧洲的,能提供按照CE医疗器械指令检测的报告或CE证书;出口到美国的,能提供美国FDA医疗器械注册证等),方可受理。
4.如受审核组织认证范围所涉及的产品不属于上述3条款范围内,但组织能够提供相关证明文件(如:监管机构的证明)也可受理认证,但必须满足上述1条的要求。
5.对于医疗器械的配件企业,需提供医疗器械采购方的采购合同。
五、资料清单
认证组织需要提交的基本资料有:
1.法律地位证明文件(如企业法人营业执照、事业单位法人代码证书、社团法人登记证等);
2.有效的资质证明、产品生产许可证、强制性产品认证证书等涉及法律法规规定的行政许可的须提交相应的行政许可证件复印件(需要时);
申请企业的产品涵盖在国家医疗器械分类目录的,申请人应提交的资质证明具体如下:
1.对于生产型企业,提供产品注册证/备案凭证、医疗器械生产企业许可证/备案凭证;
2.对于经营企业,提供医疗器械经营企业许可证/备案凭证。
3.对于产品仅供出口的企业,不需要提供以上证件。但应提供满足出口国法规的证据。
六、认证费用
可能产生的费用,包括初次认证费用(申请费、审核费、注册费等)、监督审核费用(审核费、年金等)、再认证费用(申请费、审核费、注册费等)。
费用多少要看评审工作的复杂程度等因素进行核算。如:初评、监督、复评与扩大认可领域、扩大业务领域、扩大业务范围和增加关键场所的文件审查、现场评审、见证评审、不符合验证等评审活动所发生的费用等。
七、证书样本
 |
 |
| 医疗器械质量管理认证证书 | 医疗器械质量管理认证证书(英文版) |
Copyrights © All Rights Reserved. 版权所有 -甘肃华科润知识产权代理有限公司 陇ICP备16001604号 设计制作 宏点网络






 微信
微信
 微博
微博
 在线客服
在线客服